Table of Contents
Qual é a doença de Wilson?
A doença de Wilson é um distúrbio genético que dificulta a capacidade do organismo de se ligar e transportar cobre, levando ao acúmulo de cobre no corpo. Ao longo do tempo, a deposição de cobre ocorre em vários órgãos e tecidos diferentes. O cobre na sua forma livre é tóxico e danifica os órgãos onde se acumula. Como o corpo não é capaz de controlar adequadamente o cobre, o metal se acumula no corpo a partir do momento do nascimento. No entanto, os sintomas só podem ser notados aos 5 anos de idade ou até mais tarde na vida. A doença de Wilson é rara e afeta apenas cerca de 1 em 30.000 pessoas.
Acumulação de cobre no corpo
O cobre está presente em pequenas quantidades no corpo humano. É principalmente proveniente de alimentos absorvidos no intestino. O cobre é tóxico no estado livre e está normalmente ligado a certas proteínas (ceruloplasmina), o que o torna não tóxico. A toxicidade do cobre surge quando:
- quantidades anormalmente grandes de cobre são introduzidas no corpo, ou
- quando então o corpo não pode lidar com quantidades normais de cobre apropriadamente.
A ingestão de grandes quantidades de cobre acontece por várias razões durante o curso da vida. Neste caso, é conhecido como toxicidade de cobre adquirida. Quando defeitos genéticos hereditários afetam a ligação do cobre com proteínas, seu transporte no corpo e a subsequente eliminação, ele é conhecido como toxicidade hereditária do cobre. A doença de Wilson é uma condição de toxicidade hereditária do cobre.
Deposição de Cobre nos Órgãos
O cobre é processado no fígado. Normalmente, o cobre em excesso seria excretado na bile, mas na doença de Wilson, o processamento prejudicado do cobre significa que ele se acumula no fígado. Eventualmente, o tecido do fígado é severamente danificado e as células do fígado degeneram – cirrose. O cobre no fígado se difunde na corrente sanguínea e pode se acumular em vários órgãos. O cérebro é um dos principais órgãos onde ocorre essa deposição de cobre e isso destrói o tecido cerebral.
O processo não ocorre durante a noite. Acontece ao longo de anos e décadas e essencialmente começa a partir do momento do nascimento. Os primeiros sintomas da toxicidade do cobre são vistos já aos 5 anos de idade. São principalmente sintomas de disfunção hepática, mas esses sintomas podem, às vezes, passar despercebidos. Eventualmente, os outros sintomas tornam-se evidentes, por vezes tão tarde como nos anos 30. Neste ponto, os sintomas neurológicos tornam-se evidentes e isso pode ser a primeira indicação da doença.
Estágios da Doença de Wilson
A doença de Wilson pode ser categorizada por quatro estágios à medida que a condição progride e a deposição de cobre se estende a vários órgãos do corpo.
Estágio 1
Este é o estágio durante o qual o cobre se acumula em locais específicos dentro do fígado.
Estágio 2
O cobre se espalha pelo fígado e desmaia na corrente sanguínea.
Estágio 3
Deposição de cobre a longo prazo ocorre em vários órgãos em todo o corpo. A doença progride durante esta fase, possivelmente culminando com a morte, se o tratamento médico apropriado não estiver disponível.
Estágio 4
Estabilização do desequilíbrio de cobre e reversão da doença, em certa medida, com o tratamento médico adequado.
Sinais e sintomas
Os sinais e sintomas da doença de Wilson dependem do estágio da doença e de qual órgão é infiltrado com cobre. Os sintomas do fígado são vistos pela primeira vez, mas às vezes perdidos, seguidos pelos sintomas neurológicos em um estágio posterior. No diagnóstico tardio, os sintomas hepáticos e os sintomas neurológicos são detectados ao mesmo tempo. Outros sinais e sintomas também podem estar presentes.
Sintomas hepáticos
- Icterícia – amarelecimento da pele, revestimento interno da boca e ‘brancos’ do olho (esclera).
- Retenção de líquidos, principalmente nas pernas (evidente como inchaço nos pés e tornozelos) e no abdômen (distensão abdominal).
- Fígado aumentado (hepatomegalia) ou baço (esplenomegalia) ou ambos (hepatoesplenomegalia).
- Fácil hematomas.
- Fadiga severa.
Sintomas neurológicos
- Tremores e coreia (movimentos involuntários).
- Fraqueza muscular e rigidez.
- Má coordenação.
- Dificuldade em engolir e falar.
- Mudanças na personalidade.
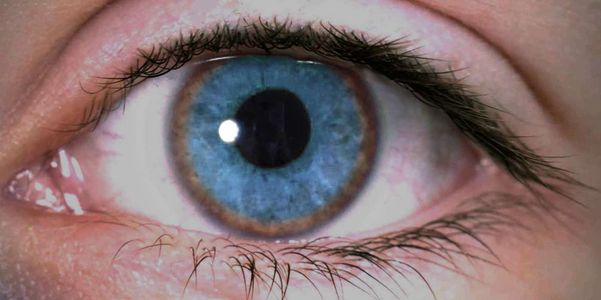
Sintomas oculares
Uma das características da doença de Wilson é o acúmulo de cobre nos olhos. Ele é visto externamente como um anel marrom-ferrugem ao redor da íris e da córnea. Isso é conhecido como anéis Kayser-Fleischer.
Foto dos anéis de Kayser-Fleischer da Wikimedia Commons
Outros sintomas
A toxicidade do cobre tem uma ampla gama de efeitos em diferentes órgãos e sistemas, não apenas no fígado e no sistema nervoso central (SNC).
- Baixos níveis de eritrócitos e hemoglobina (anemia).
- Distúrbios de coagulação.
- Baixa contagem de plaquetas (trombocitopenia).
- Contagem de glóbulos brancos baixa (leucopenia).
- Proteína na urina (proteinúria).
- Níveis elevados de ácido úrico na urina (uricosúria).
- Inflamação das articulações (artrite).
- Osteoporose precoce.
Causas da toxicidade de cobre
A toxicidade do cobre pode ser adquirida quando grandes quantidades de cobre são ingeridas ou absorvidas pela pele. Mesmo usando panelas de cobre para cozinhar pode ser uma fonte de toxicidade de cobre. Na doença de Wilson, a causa da toxicidade do cobre é devido a um defeito herdado na forma como o corpo manipula o cobre. O corpo retém muito pouco cobre e cobre que é absorvido pelo intestino é geralmente eliminado na bile. Com a doença de Wilson existe um defeito no gene ATP7B que afeta a capacidade do fígado de excretar cobre na bile. O cobre sobrecarrega o fígado e, eventualmente, se espalha por todo o corpo.
Fatores de risco
A doença de Wilson é herdada. Genes mutados são passados do pai para o filho. É um distúrbio hereditário autossômico recessivo que significa que duas cópias defeituosas do gene são necessárias para a doença surgir – uma cópia de cada genitor. No entanto, os pais de crianças com doença de Wilson podem não ter a doença, apesar de portarem os genes defeituosos (portadores).
Testes e Diagnóstico
Além dos anéis característicos de marrom-ferrugem a marrom-dourado ao redor da íris (anéis de Kayser-Fleischer), a doença de Wilson é difícil de diagnosticar apenas nos sinais e sintomas. Mesmo os anéis de Kayser-Fleischer não são óbvios e só são detectados durante um exame oftalmológico. Em geral, a doença de Wilson é uma condição rara e muitas outras doenças mais comuns têm características clínicas semelhantes. Por isso, vários testes de laboratório são necessários.
Fluidos corporais
O sangue e a urina podem ser testados para detectar a quantidade de cobre dentro dele. Outros exames de sangue auxiliam na avaliação da função hepática ou dos níveis da proteína (ceruloplasmina) que transporta cobre no sangue. Enquanto os níveis de cobre no sangue são elevados com a doença de Wilson, o nível de ceruloplasmina é geralmente baixo.
Biópsia hepática
Uma pequena amostra de tecido hepático pode ser coletada (biópsia) e enviada para testes laboratoriais para avaliar os níveis de cobre dentro do tecido hepático. O exame microscópico também revelará o grau de dano hepático.
Genes
O teste genético para detectar os genes defeituosos que causam a doença de Wilson ajuda a confirmar o diagnóstico. Também é útil na triagem dos irmãos de um paciente com doença de Wilson e, possivelmente, começar com o tratamento precoce em pacientes que ainda não apresentaram sintomas notáveis da doença.
Tratamento da Doença de Wilson
A principal abordagem para tratar a doença de Wilson é remover o excesso de cobre no organismo (terapia de quelação) e reduzir a ingestão de cobre através da dieta.
Terapia Quelante
Agentes quelantes ajudam a remover o excesso de cobre no corpo, estimulando os órgãos a liberar cobre na corrente sanguínea. Embora o cobre não possa ser excretado em grau suficiente através da bile, o rim é capaz de filtrar o cobre e passá-lo pela urina. A penicilamina é o principal agente quelante utilizado na doença de Wilson. Se isso não for bem tolerado, o cloridrato de trientina é usado como alternativa.
Agentes quelantes podem precisar ser usados por toda a vida, mas em doses baixas (terapia de manutenção), uma vez que o excesso de cobre é excretado do sistema.
Zinco
Às vezes, o acetato de zinco é prescrito quando a terapia de quelação não é viável por longos períodos. O acetato de zinco previne o acúmulo de cobre nos tecidos e é uma opção, juntamente com uma dieta pobre em cobre, em pacientes que não toleram a terapia de quelação. É importante notar que o acetato de zinco não deve ser usado com agentes quelantes.
Alimentos ricos em cobre
O cobre está presente em quantidades mínimas em muitos alimentos. Não é possível evitar completamente o cobre dietético. No entanto, alimentos ricos em cobre devem ser evitados ou consumidos com moderação. Estes alimentos que são abundantes em cobre incluem:
- abacates
- feijão (seco)
- castanha de caju
- cacau e chocolate
- fruta (seca)
- lentilhas (secas)
- fígado
- cogumelos
- nozes
- ervilhas (secas)
- marisco
- suco de vegetais ou outros concentrados vegetais
Suplementos vitamínicos e minerais contendo cobre devem ser evitados.



